On September 29, 2023, and as widely anticipated, the Food and Drug Administration (FDA) announced the publication of a proposed rule to “clarify” that LDTs are medical devices subject to FDA regulation.
LDTs are diagnostic tests that are developed and offered by high-complexity laboratories certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA). While FDA has asserted that it has authority to regulate LDTs as medical devices, it has never broadly exercised that authority. In the proposed rule, FDA seeks to amend its regulation defining “in vitro diagnostic products” (IVDs) to add the words “including when the manufacturer of these products is a laboratory.”
While the proposed new regulatory text is only 10 words, the majority of the regulatory preamble is focused on FDA’s justification for issuing the proposed rule and its claimed legal basis for doing so. This Client Alert does not address FDA’s claimed policy justifications—including its characterization of “problematic LDTs”—or FDA’s legal authority to issue the rule. Both topics are highly controversial and will undoubtedly be contested by stakeholders. Instead, this Alert is focused on the substance of the proposed policy—a five-stage “phase out” of FDA’s enforcement discretion policy for LDTs over four years. As described below, the details are slim, and many questions remain unanswered.
Covington has been deeply involved in the debate over FDA’s authority in this space, including a long-standing representation of the American Clinical Laboratory Association and submission of a citizen petition in 2013 challenging FDA’s authority to regulate LDTs under its device authorities. In addition, Covington regularly represents diagnostic companies with respect to FDA and CLIA regulation.
Brief Background: History of LDT Regulation
FDA has asserted that LDTs qualify as medical devices, but the Agency has not actively regulated such tests or resolved significant questions about its authority to do so. This means that the Agency has not attempted to require compliance with the Federal Food, Drug & Cosmetic Act (FDCA)—with the exception of certain test categories, such as direct-to-consumer tests, some pharmacogenomic (PGx) tests, and tests that respond to public health emergencies. Meanwhile, many in the laboratory community and others have long taken the position that FDA does not have authority under the FDCA to regulate LDTs.
At the same time, over the past several years, Congress has worked with key stakeholders on legislative proposals for regulating diagnostics, including LDTs. Most notable among these proposals was the Verifying Accurate Leading-edge IVCT Development (VALID) Act, which was introduced in both the House and Senate, closely negotiated for months, and nearly passed at the end of 2022 in the Consolidated Appropriations Act of 2023. Ultimately, the bill was not enacted, and in the spring of 2023, FDA announced its intention to initiate rulemaking for LDTs.
Discussion
As noted above, the text of the proposed rule is short: 10 words asserting that IVDs include products manufactured by a laboratory. The stages for the proposed “phase out” of FDA’s decades-long enforcement discretion policy is summarized in a single page, with additional justification and some detail over the following 9 pages. The scope and timeline for the phase out is summarized below.
Scope of Proposed Phaseout Policy
With regard to scope of the phaseout policy, FDA states that although its traditional understanding of LDTs was limited to diagnostics designed, manufactured, and used within a single CLIA-certified, high-complexity laboratory, the proposed “phaseout” policy would be broader, and include “IVDs that are manufactured and offered as LDTs” by CLIA-certified, high-complexity laboratories (emphasis in original). The proposed rule does not clarify what it means to be “manufactured and offered as LDTs.”
FDA also proposes that enforcement discretion would continue to apply to certain types of tests, which would not be subject to the phaseout policy. These tests include “1976-type LDTs” (tests that use manual techniques without automation), Human Leukocyte Antigen tests, tests for forensic (law enforcement) purposes, and tests intended for public health surveillance, each category subject to certain conditions described in the preamble.
Finally, FDA asserts that certain types of tests were not subject to its enforcement discretion approach for LDTs and accordingly proposes that the phaseout policy would not apply to these tests. Rather, such tests would continue to be subject to FDA regulation as devices. These tests include:
(1) Direct-to-consumer tests;
(2) Tests intended for emergency use under section 564 of the FD&C Act; and
(3) Tests intended as blood donor screening or human cells, tissues, and cellular and tissue based products (HCT/Ps) donor screening tests required for infectious disease testing or for determination of blood group and Rh factors.
Timeline for Phaseout Policy
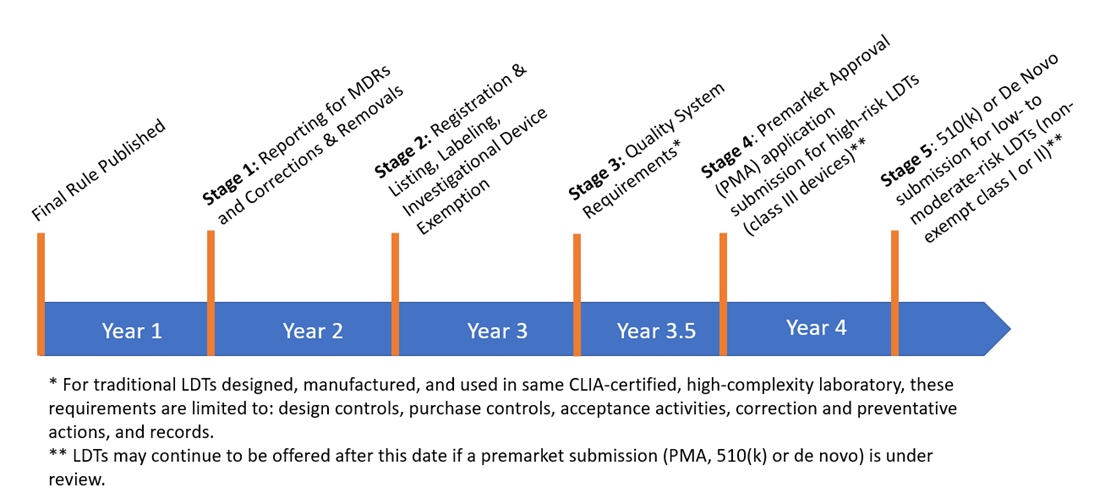
The phaseout policy for all in-scope tests would consist of five stages, with each stage marking the end of FDA’s general enforcement discretion policy for different regulatory requirements. The five stages each begin at a certain time “after FDA publishes a final phaseout policy,” which is intended to be published in the preamble to the final rule, as follows:
- Medical device reporting (MDR) (21 CFR Part 803) and corrections and removal reporting (21 CFR Part 806) – 1 year after final phaseout policy published.
- Registration and listing (21 CFR Part 807), labeling (21 CFR Parts 801 and 809, subpart B), and investigational use requirements (21 CFR Part 812) – 2 years after final phaseout policy published.
- Quality system (QS) requirements (21 CFR Part 820)[1] – 3 years after final phaseout policy published. However, for traditional LDTs, i.e., LDTs that are designed, manufactured, and performed within the same CLIA-certified, high-complexity laboratory, only the following quality requirements would apply: design controls (21 CFR 820.30), purchasing controls (21 CFR 820.50), acceptance activities (21 CFR 820.80 and 21 CFR 820.86), corrective and preventative actions (21 CFR 820.100), and records (21 CFR Part 820, subpart M).
- Submission of premarket approval (PMA) applications for high-risk tests, i.e., class III devices – 3.5 years after final phaseout policy published, but no earlier than October 1, 2027. LDTs generally could continue to be offered after this date as long as a PMA is under review.
- Submission of 510(k) premarket notifications and de novo requests for moderate- and low-risk tests (i.e., non-exempt class I and II devices) – 4 years after final phaseout policy published, but no earlier than April 1, 2028. LDTs generally could continue to be offered after this date as long as a premarket submission is under review.[2]
A timeline of the phaseout policy is summarized below.
Requests for Comment:
While stakeholders may submit comments on any topic associated with the proposed rule, FDA specifically requested comments on the following topics:
- Whether all or some LDTs should be “grandfathered” from premarket review or QSR requirements;
- Whether a longer phaseout period should be available for LDTs offered by small laboratories;
- Whether a different policy should apply for LDTs offered by academic medical centers (AMCs); and
- Whether programs such as New York State’s Department of Health Clinical Laboratory Evaluation Program (NYSDOH CLEP) or those within the Veterans Health Administration (VHA) could be leveraged such that it would be appropriate to continue FDA’s general enforcement discretion for tests in compliance with such programs.
Open Questions
Following publication of the proposed rule many fundamental questions remain unanswered. These include (but are in no way limited to):
- Are LDTs devices under the FDCA, or does FDA lack legal authority to regulate LDTs?
- Even if FDA had authority to regulate LDTs as devices, would such regulation advance the public health as FDA asserts, or be unduly burdensome without corresponding benefits?
- With regard to scope of the proposed rule, what does it mean to be “manufactured and offered” as an LDT?
- How would laboratories comply with certain FDA requirements that are inconsistent with the nature of LDTs? As only one example of many, how would a laboratory comply with FDA labeling requirements for a test that is never assembled and distributed?
- Will FDA publish additional guidance, whether in the preamble to the final rule or elsewhere, with additional detail regarding implementation of the proposed phaseout policy?
- How does this proposed phaseout policy align with other FDA programs? For example, FDA recently launched a pilot program for companion diagnostic tests used with oncology drug products. Under the pilot program, FDA would publish recommended minimum performance characteristics for tests intended to be used as companion diagnostics with a particular drug, and the clear implication is that LDTs meeting these performance characteristics would be subject to enforcement discretion even when they make companion diagnostic claims for the drug product. How would LDTs offered under the pilot program be affected by the proposed phaseout policy?
- What would FDA regulation of LDTs mean for manufacturers of instruments, reagents, and other tools used by laboratories, including those distributed as research use only (RUO) products?
- How will the proposed rule apply to companies offering bioinformatics or other software utilized by laboratories offering LDTs?
- Will FDA issue any new or updated policies for in vitro diagnostics in conjunction with the proposed phaseout policy for LDTs?
- Does FDA have adequate resources to manage implementation of the phaseout policy, if finalized?
Next Steps
The proposed rule is scheduled to be published in the Federal Register on Tuesday, October 3, 2023, and comments may be submitted until December 4, 2023.
With respect to finalizing the rule, the proposed rule states that FDA anticipates that stage 4 of the proposed phaseout policy could begin as early as October 1, 2027 to align with the beginning of the next user fee cycle for devices. To meet this timeline, FDA would need to publish the final rule as early as April 1, 2024, a mere 5 months after the planned close of the comment period. This is a very aggressive timeline for finalizing any rulemaking, and particularly one that would alter the regulatory landscape for an entire field of tests.
Moreover, some stakeholders have already announced that they will request additional time to comment on the proposed rule, which could make the timeline for finalizing the rule even more challenging.
If you have any questions concerning the material discussed in this client alert, please contact the members of our Food, Drugs, and Devices practice.
[1] The proposed rule acknowledges that on February 23, 2022, FDA proposed to amend the QSR to align more closely with international consensus standards for devices, currently set forth in ISO 13485:2016, and because FDA intends to finalize these amendments expeditiously, the amended QSR may be in effect before the proposed beginning of stage 3.
[2] The proposed rule anticipates that laboratories may seek to utilize FDA’s Third Party review program for these premarket submissions, and suggests that CLIA accreditation organizations potentially could become Third Party reviewers under such program.
 Back
Back











